
DNA甲基化图谱分析,尤其是在基因组中检测5-甲基胞嘧啶(5mC)和5-羟甲基胞嘧啶(5hmC),是至关重要的,甲基化修饰会影响基因的表达。通常,转录起始位点附近的低甲基化水平与较高的转录水平有关,而调节区域内高水平胞嘧啶修饰的基因则出现较低转录水平。完整和准确的甲基化图谱分析在许多领域都很重要:包括癌症等疾病的研究、胚胎发育监测和农业植物研究。然而,现有的甲基化图谱分析技术存在明显的缺陷。
全基因组重亚硫酸盐测序(WGBS)长期以来一直是甲基化图谱分析的金标准,但重亚硫酸盐的化学反应会破坏和降解DNA,导致DNA断裂和丢失。此外,重亚硫酸盐文库表现出明显的GC偏嗜性,并在甲基化区域尤为显著。
测序中国近日获悉,为了克服这些局限性,NEB®公司(New England Biolabs®)成功开发了一种基于酶学转化、能同时检测5mC和5hmC的新方法——NEBNext酶学转化法甲基化建库试剂盒 (EM-seq™)。
NEB介绍,高效的酶法转化可更大程度地减少对DNA的损伤,结合该公司提供的NEBNext Ultra™ II文库制备流程,最终产生的高质量文库可以从有限的测序数据中更灵敏地检测到5mC和5hmC。EM-seq方法得到的结果与WGBS得到的转化序列相同,因此可以使用相同的数据分析流程。
EM-seq流程的第一步,与WGBS方法相同,都是先从打断DNA开始构建文库。EM-seq的标准起始量为10~200ng的打断后的DNA,优化后的流程也可用于高达500ng的起始量。紧接着是两步酶法转化,目的是将未甲基化的胞嘧啶与5mC和5hmC区分开来。最后,在上机测序前对文库进行PCR扩增。EM-seq的两步酶促转化与重亚硫酸盐转化如图1所示。
图1:EM-seq和重亚硫酸盐转化的方法原理
相较于过于损伤DNA的重亚硫酸盐处理,EM-seq流程对DNA更为温和,更大程度地减少了对DNA的损伤。结果显示,EM-seq转化后的DNA比重亚硫酸盐转化后的DNA更为完整,得到的文库中拥有更多更长的插入片段,如图2所示。最终能获得更长的序列和更高的比对率,同时尽可能地降低测序成本。
图 2:NEBNext酶学转化法甲基化文库可获得更长的插入片段
采用Covaris® S2仪器将50ng人NA12878 基因组DNA打断至300bp,同时作为EM-seq和WGBS建库的起始样本。对于WGBS方法,使用NEBNext Ultra II DNA进行建库,随后采用Zymo Research EZ DNA Methylation-Gold™试剂盒进行重亚硫酸盐转化。两个文库均使用Illumina MiSeq(2 X 76 bases)测序,片段大小采用Picard 2.18.14测定。图中绘制了每个插入片段出现的频率标准化后的数据,结果如图:EM-seq建库后的插入片段比 WGBS 方法得到的插入片段更长,说明:酶学转化法不会对DNA造成损伤,而重亚硫酸盐处理的DNA有严重损伤。
重亚硫酸盐转化引起的DNA损伤、断裂和丢失降低了其文库扩增后的产量。相反,在EM-seq流程中更温和的转化方式可保证获得高质量的DNA文库,继而进行高效扩增。因此,相较于WGBS,EM-seq可用更少的PCR循环实现更高的文库产量(图3)。
图3:相较于WGBS,EM-seq能在更少的PCR循环下得到更高的文库产量。
采用Covaris® S2仪器将10ng、50ng和200ng不同起始量的人NA12878基因组DNA打断至300 bp,同时作为EM-seq和WGBS建库的起始样本。对于WGBS方法,使用NEBNext Ultra II DNA进行建库,随后采用Zymo Research EZ DNA Methylation-Gold试剂盒进行重亚硫酸盐转化。上述所有起始量,EM-seq都能使用更少的PCR循环得到更高的文库产量,表明EM-seq显著减少了WGBS方法中的DNA损失。误差条表示标准差。
虽然文库产量是测序成功的要素,但文库质量也同样至关重要。一个高质量文库应能真实反映原始样本情况,包括均一的GC覆盖度。
由于重亚硫酸盐转化时会损伤非甲基化胞嘧啶,而非甲基化胞嘧啶占所有胞嘧啶的绝大部分,因此这种导致损伤的转化方式会对GC比例造成影响。这种特定的损伤、断裂和丢失导致重亚硫酸盐转化后的文库在GC测序不足,而在AT过度测序。相比之下,EM-seq文库显示了更均一的 GC 覆盖度,结果表明:EM-seq对DNA的损伤更少,更能真实反映原始样本的情况(图 4)。
图4:EM-seq能实现更卓越的GC均一覆盖度
采用Covaris® S2仪器将10ng、50ng和200ng不同起始量的人NA12878基因组DNA打断至300bp,同时作为EM-seq和WGBS建库的起始样本。对于WGBS方法,使用NEBNext Ultra II DNA进行建库,随后采用Zymo Research EZ DNA Methylation-Gold™试剂盒进行重亚硫酸盐转化。两个文库均使用Illumina NovaSeq® 6000(2X 100 bases)测序,使用bwa-meth0.2.2将测序数据与hg38进行比对。使用 Picard 2.17.2 计算GC覆盖度,图中显示不同GC含量时(0-100%),标准化后覆盖度的分布情况。结果显示:EM-Seq 文库显著提高GC覆盖度的均一性,无AT过度测序,也无GC测序不足,后两项都是WGBS文库的典型缺陷。
分析两种转化方法在不同测序深度下检测到的CpG位点数量,可以比较EM-seq 和WGBS对CpG位点的检测灵敏度。图5显示:在10ng、50ng和200ng三种起始量下,分析相同的数据量(3.24 亿),EM-seq文库能比重亚硫酸盐文库检测到更多的CpG位点,并且在更低测序深度下,EM-seq也能检测到更多的CpG位点。例如,用EM-seq和WGBS两种方法在不同起始量情况下,至少1X和8X测序深度下检测到的独有和共有的CpG位点数量,EM-seq在至少1X测序深度下比WGBS多检测到20%以上的CpG位点。而在至少8X测序深度下,CpG位点覆盖度差异增加至2倍。
图5:相较于WGBS,EM-seq在更低测序深度能检测到更多的CpG位点。
使用bwa-meth 0.2.2将测序数据与hg38进行比对。通过分析3.24亿双端数据得到EM-seq和WGBS文库的CpG位点覆盖度,其中每条链都独立计数,最终得到最多5600万个可能的CpG位点。结果显示:EM-seq在更低测序深度能检测到更多的CpG位点。
图6显示了EM-seq和WGBS两种方法在不同起始量,不同测序深度,采用相同测序数据检测到的独有和共有的CpG位点,同样表明EM-seq能够检测到更多的CpG位点,并且这种差异在提高测序深度后更为显著(图 6)。在至少1X测序深度下,使用10、50和200ng作为起始样本,EM-seq能检测到约5400万个CpG位点,但WGBS只识别了约3600万至4600万个CpG位点。在至少8X测序深度下,EM-seq检测到的CpG位点数在1100万至1660万,而WGBS仅检测到160万至710万。结果表明,与WGBS相比,EM-seq能从更少的数据量中挖掘到更多的相关信息(CpG 覆盖度)。
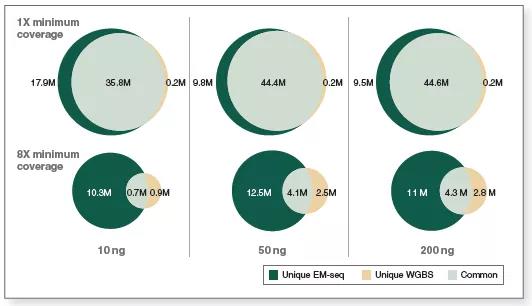

图6:相较于WGBS,EM-seq在更低的测序深度情况下能检测到更多的CpG位点
采用Covaris® S2仪器将10ng、50ng和200ng不同起始量的人NA12878基因组DNA打断至300bp,同时作为EM-seq和WGBS建库的起始样本。对于WGBS方法,使用NEBNext Ultra II DNA进行建库,随后采用Zymo Research EZ DNA Methylation-Gold™试剂盒进行重亚硫酸盐转化。两个文库均使用Illumina NovaSeq® 6000(2X 100 bases)测序,使用bwa-meth 0.2.2将测序数据与hg38进行比对。通过分析 3.24 亿双端数据得到EM-seq和WGBS文库的CpG位点覆盖度。
图中显示了EM-seq和WGBS两种方法在不同起始量,至少1X 和8X测序深度下检测到的独有和共有的CpG位点。EM-seq在至少1X测序深度下比WGBS多检测到20%以上的CpG位点。而在至少8X测序深度下,CpG位点覆盖度差异增加至2 倍。
不同起始量和不同方法所产生的文库相关性,是评估一致性和高性能的有效参数。图7显示了采用EM-seq和WGBS两种方法,在不同起始量下,检测了12个文库各 2100 万个CpG位点间的 CpG 甲基化相关性。EM-seq文库在不同起始量之间的相关性都很高,表明甲基化检测灵敏度不会随着起始量的减少而降低。相比之下,WGBS文库之间的相关性最低。只有在较高的起始量下,EM-seq和WGBS文库之间的相关性才最高。综上所述,表明EM-seq具有更高的一致性。
图 7:相较于WGBS,EM-seq文库具有更高的CpG相关性
采用methyKit绘制10、50和200ng起始量下EM-seq和WGBS文库之间的相关性,最小覆盖度为1×(所有文库均使用2100万个 CpG 位点)。EM-seq文库在不同起始量之间的相关性都很高,Pearson相关系数在0.82到0.86之间。而WGBS文库之间的相关性仅在0.71到0.8之间。起始量在50ng和100ng时,EM-seq和WGBS之间的相关性最高,而在起始量降至10ng时,两者的相关性最低。结果表明:比较EM-seq和WGBS数据后,发现EM-seq具有更高的一致性。
在基因表达中,转录起始位点区域(TSS)通常是非甲基化的,而在基因较低水平表达时,调控区域的胞嘧啶甲基化水平较高。因此,能够准确检测转录调控元件是至关重要的。然而,如上文所述,WGBS数据始终具有较低的含C二核苷酸表达问题,最终可能导致重要转录调控元件周围的CpG数据丢失。
图8显示了转录起始位点(TSS)周围的CpG覆盖度和甲基化情况。在TSS周围4 kb区域中,EM-seq得到的CpG覆盖度显著高于WGBS,并且更均一,也没有在TSS处出现CpG覆盖度下降的低峰,而WGBS在TSS处表现明显低峰。在8X条件下,EM-seq获得的甲基化信息也与预期的甲基化模式更为一致,越接近TSS区域,甲基化水平越低。
图 8:EM-seq在检测转录起始位点周围的 CpG 甲基化方面优于WGBS
图中检测了不同起始量下转录起始位点(TSS)周围CpG位点甲基化情况。TSS周围的4kb被分成了400个10bp,在该区域内CpG位点被覆盖 8×及以上的才能被用来评估甲基化水平。A:结果表示:EM-seq在TSS区域有更高且更均一的覆盖度。B:图中显示了EM-seq和WGBS在8×覆盖度下,CpG位点的平均甲基化百分比。EM-seq在TSS区域的数据更能代表预期的甲基化模式:TSS区域的甲基化水平最低,而在+/-2 kb处甲基化水平增高。
结 语
虽然重亚硫酸盐测序是研究 DNA 甲基化的金标准,但这种转化方式会对DNA 造成损伤,导致DNA断裂、丢失和GC偏嗜。NEBNext酶学转化法甲基化建库试剂盒提供了一种酶学方法代替全基因组重亚硫酸盐处理DNA (WGBS) 的方法,并结合高效流程化的建库步骤,适用于Illumina平台测序。
综上所述,高效的EM-seq酶促转化可更大程度地减少对DNA的损伤,结合提供的NEBNext Ultra II文库制备流程,最终制备的高质量文库可以从有限的测序数据中更灵敏的检测到5-mC和5-hmC。
点击下方图片
可申请参与“NEB 甲基化测序建库体验星标计划”
客服热线: 400-811-2220 / 400-690-3366
本文由 SEQ.CN 作者:白云 发表,转载请注明来源!

