
近日,卫生部临床检验中心(以下简称“临检中心”)公布《 2018 年国际肿瘤游离DNA(ctDNA)基因突变检测室间质量评价调查活动结果报告 》。
肿瘤游离 DNA(ctDNA)基因突变检测对肿瘤靶向治疗、早期治疗应答评估和耐药监测的实时评估等都具有一定的临床应用价值。由于组织样本的局限性,临床上逐渐开始使用患者血浆中的游离 DNA 进行肿瘤基因突变的检测。为了解临床实验室 ctDNA 基因突变检测的开展现状及质量状况,临检中心开展了该项目室间质量评价的预研, 要求各临床实验室使用日常所用试剂和程序进行检测。
本次质评由卫生部临床检验中心和澳大利亚皇家病理质控中心(the Royal College of Pathologists of Australasia Quality Assurance Programs, RCPAQAP)联合开展,所有质评样本均为卫生部临床检验中心制备。 本次参评单位包括中国和澳大利亚开展肿瘤游离 DNA 基因突变检测的实验室。
我国共计 116 家实验室参加了本次室间质评, 部分实验室回报的结果为无效结果, 包括以下情况:未回报或者未按时回报结果, 结果回报不完整(缺少 run report 或者 runParameters 文件、缺少结果报告单、 缺少回报表等), 回报项目与报名时方法学不符, 使用同一台测序仪或者同一个 flowcell 等。对于无效结果,不进行评价。
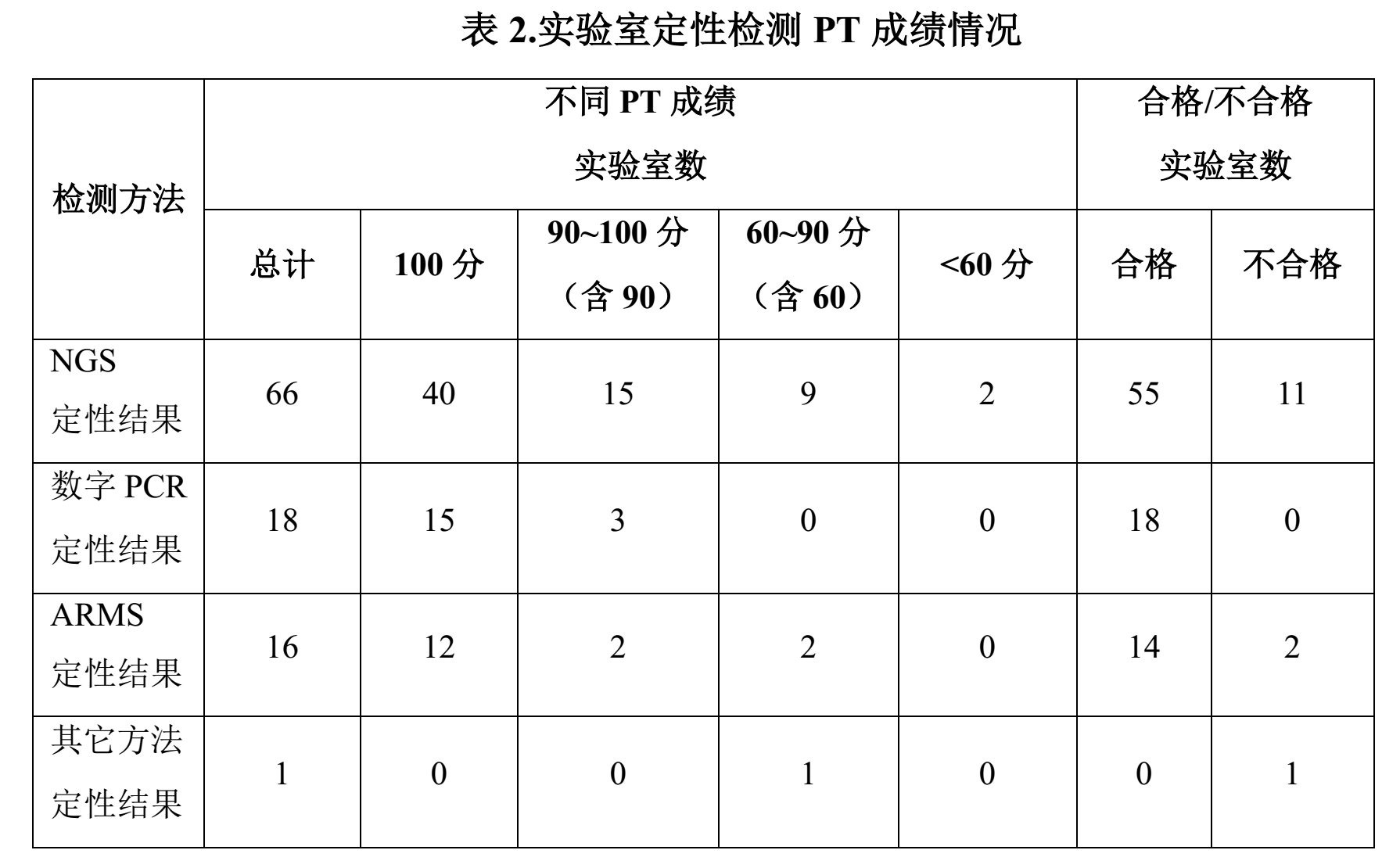
本次共计收到 101个有效结果,满分实验室60余个,满分率66.3%(部分实验室回报 2种或 2种以上方法的结果),其中 NGS 方法 66 个、满分率60.6%; 数字 PCR 法 18 个,满分率83.3%; ARMS 法 16 个、 满分率75%。其它方法 1 个。 详细名单见下表,(排名不分先后)
澳大利亚共计 12 家实验室参加了本次室间质评, 有效结果为 8个。澳大利亚评价结果由澳大利亚皇家病理质控中心公布。
1. 基本情况
共发放 8 个质评样本,编号分别为 1801、 1802、 1803、 1804、 1805、 1806、1807、 1808。 为已提取的血浆游离 DNA,游离 DNA 浓度为 3~5ng/µl,样本量为25µl。 1801~1808 号样本均提供临床信息。
18NC 号样本,为已提取的人基因 DNA,浓度 30~50ng/µl,样本量 25µl。
2. 质评样本突变等位基因百分比的定值
由于目前没有基因突变等位基因百分比定量的“金标准”方法,预期结果中采用如下方法来确定等位基因百分比:
(1)质评样本制备时,有预期浓度范围;
(2) 根据临床实验室回报的 NGS 及 dPCR 结果, 计算每个待评价样本中各突变位点所有实验室检测突变频率(mutant allele frequencies, MAF) 结果的修正均值(X)和标准差(SD)(取 75%CI 的结果)。
(3) 修正均值在预期浓度范围内,即为预期的基因突变等位基因百分比。
3.预期结果
本次评价的基因突变预期结果, 具体信息见下表 :
1. 本次质评的检测项目是肿瘤游离 DNA(ctDNA)基因突变检测。
2. 本次室间质量评价调查活动在向每个参加实验室发送检测样本的同时,附有给实验室的活动说明“2018 年国际肿瘤游离 DNA(ctDNA)基因突变检测室间质量评价调查活动安排及注意事项”、 “2018 年国际肿瘤游离 DNA(ctDNA)基因突变 ARMS 和数字 PCR 检测室间质量评价调查活动回报表”以及“2018 年国
际肿瘤游离 DNA(ctDNA)基因突变高通量测序检测室间质量评价调查活动回报表。
“肿瘤游离 DNA(ctDNA)基因突变检测”室间质量评价的评价原则如下:
由于本次质评的评价包括两个部分
1. 基因突变的定性结果评价
(1) 假阴性结果: 在检测范围内、检测下限以上的基因突变均要求正确报
告,未报告即为假阴性结果,每个假阴性结果扣 10 分。
(2) 假阳性结果: 预期结果以外的基因突变结果,均为假阳性结果,每个
假阳性结果扣 10 分。
(3) Cutoff 值以下结果直接报告为阳性, 扣 10 分。
(4) PT 成绩计算:累计扣分, 90 分或 90 分以上为合格。
2. 基因突变的定量结果评价 仅在实验室基因突变定性结果合格的情况下,评价所报告的等位基因百分比结果;如果定性结果不合格,则不进行定量结果评价。
(1) 计算实验室报告的每个突变等位基因百分比的 z 值;
z=(x─X) /SD
注: x 为某实验室报告的某个突变等位基因百分比的 MAF 结果; X 为所有实验室对该突变位点检测 MAF 结果的修正均值, 即表 1 中“突变频率”; SD 为所有实验室对该突变位点检测 MAF 结果的修正标准差, 即表 1 中 SD 值;
(2) z值介于 2.0和-2.0之间为“在允许范围内”,判定为可接受结果;z值>2.0或 z 值<-2.0 为“不在允许范围内”,判定为不可接受结果。 一个不可接受结果扣10 分。
(3) PT 成绩计算: 不计算 PT 成绩,实验室自我教育。
定性 PT 成绩合格的实验室,发放定性质评合格证书。 PT 成绩不合格的实验室,发放质评参加证书; 无效结果,不评价,不发放证书。
本次质评实验室 PT 成绩情况见下表 :
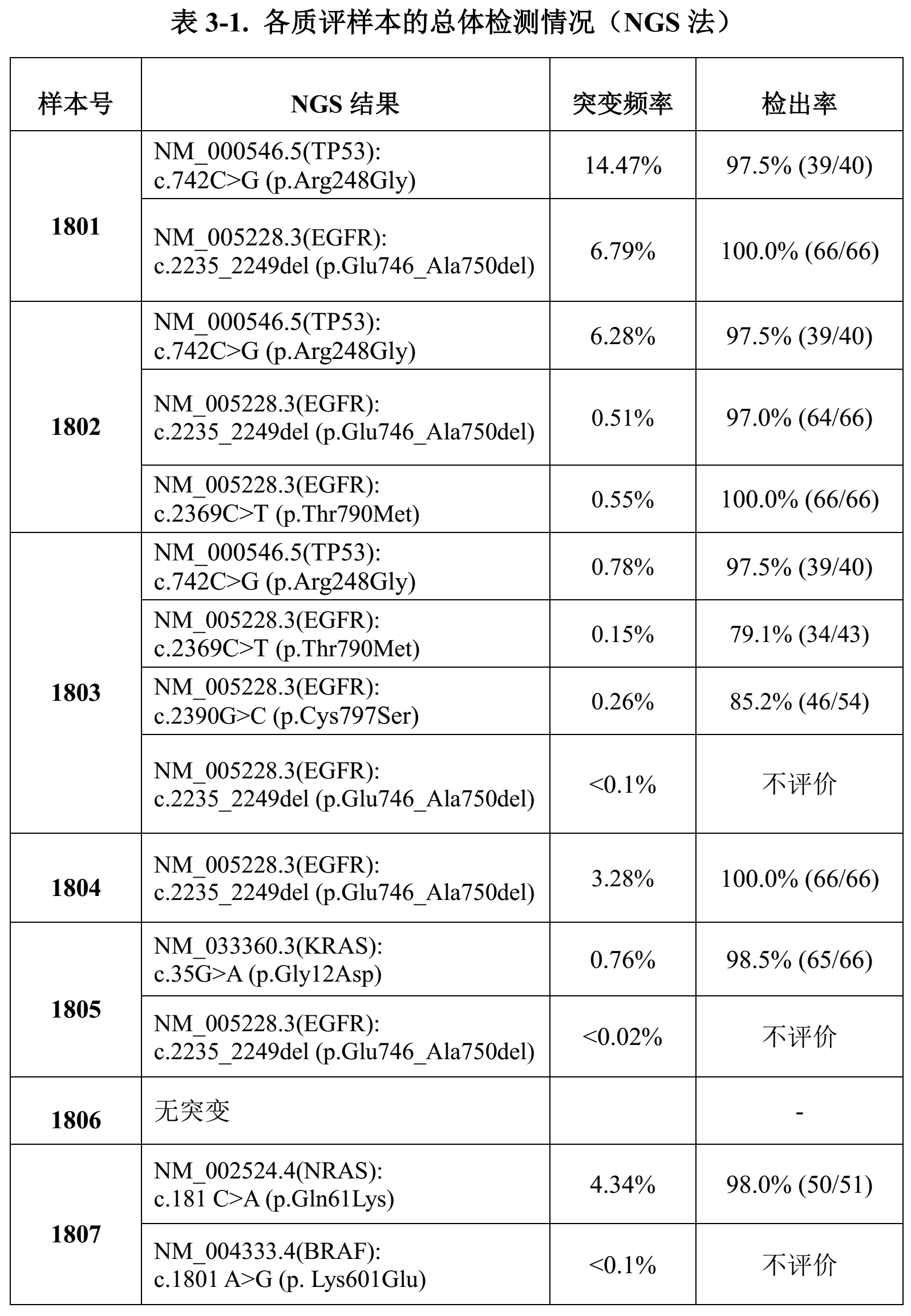
各质评样本的总体检测情况见下表 ,本次共计报告了 21 个假阳性突变结果 。


本次质评面向中国和澳大利亚开展肿瘤游离 DNA 基因突变的实验室,从2019 年开始,将接受全世界各国实验室的报名,并将继续与 RCPAQAP 在该领域进行促进实验室检测标准化的合作。
本次质评评价的是实验室进行 ctDNA 基因突变检测能力。 室间质评样本突变等位基因百分比约在 0.01%~15%, 由于绝大多数的实验室宣称的检测下限在0.1%~0.8%,因此对等位基因百分比<0.1%的样本不进行评价。
1. 部分实验室对检测的质量保证理念极其薄弱 从回报结果来看, 参评实验室使用的方法多数为自建方法,如果是自建方法,实验室应该建立试剂配制、检测过程的标准操作程序,并进行方法优化和性能确认;如果是使用批准的商品化试剂,应该进行性能验证。 但是部分实验室完全没有标准操作程序和性能确认的概念。 例如部分实验室对 cutoff 值以下的突变也直接报告。 仅有一家实验室对cutoff 值以下的的突变在报告时进行了说明,“手工审核数据可信且具有强的临床意义,则在报告中用特殊说明来详细描述该突变的具体情况”。 在这里也向国内参评实验室介绍一下澳大利亚某参评实验室在遇到 cutoff 值以下,怀疑可能为阳性样本的回报方式。该实验室采用 NGS 进行检测, cutoff 值为 0.5%,当在 1808号样本检测到 0.40%的 NM_004333.4(BRAF):c.1799T>A, p.(Val600Glu)突变时,实 验 室 报 告 :“ 我 们 检 测 到 0.40% 的 NM_004333.4(BRAF):c.1799T>A,p.(Val600Glu)突变,但是该突变低于本实验室的检测判定阈值,我们通过等位基因特异性 PCR 方法进行了确认。”
本次质评要求在检测范围内、检测下限以上的基因突变均要求正确报告,这是一个较低而且符合临床实验
室实际检测情况的要求。但是不少实验室对自建方法的检测范围不明确,对宣称的检测范围内各浓度水平的突变(如 TP53 和 NRAS)均未检出, 由于这些突变等位基因百分比有的远高于 1% ,因此这些实验室应该不具有对这两种突变的检测能力,与其宣称的检测范围严重不符。部分实验室所宣称的检测下限,实际检测能力也无法达到,实验室需要明确检测下限是可以达到 95%以上可信检出的界限,不是偶尔一次或者 50%能检出的浓度。例如实验室宣称检测下限为 0.2%,实际上 0.5%浓度的突变也未能检出。 这些都进一步反映出实验室对性能确认/性能验证方面存在很大欠缺。采用批准商品试剂的实验室同样存在很多假阴性的问题,对于肿瘤游离 DNA 基因突变检测来说,由于常常突变浓度较低,实验室应该更加重视对检测的性能验证。此外, 血浆游离 DNA 提取对肿瘤游离 DNA 检测非常重要, 本质评样本没有覆盖提取过程, 实验室应对提取效率进行充分性能验证。
本次质评不接受外送测序的实验室报名,要求全部流程均在本实验室完成,如果交给测序服务公司测序,如何保证检测质量?如何进行仪器性能的确认、检测性能的确认,如何控制这一环节的检测过程?
总之,本次室间质评调查一定程度上反映了目前肿瘤游离 DNA 基因突变检测的现状以及存在问题,这些也是目前实验室分子检测的共同问题。 我国肿瘤游离 DNA 基因突变检测的规模和投入较大,但是实验室自建方法在我国相对起步较晚,实验室人员需要明确, 只有分析前中后过程中严格的质量保证,自建方法
才有可能进行临床应用。性能确认非常重要, 凭借经验所建立的方法, 应视为可能报告错误结果的方法,而不是可靠的方法。性能确认的过程,就是确认哪些方面的结果是可靠的,哪些方面的结果是不可靠的, 对不可靠的情况不纳入检测范围或者建立补充实验程序。
感谢厦门艾德医学检验所、南京世和医学检验所、 Thermo Fisher 公司为本次室间质评样本制备过程中完成的部分协助验证工作。
国际肿瘤游离 DNA(ctDNA)基因突变检测室间质评证书将于 2018 年 7 月31 日前发放。
本文由 SEQ.CN 作者:戴胜 发表,转载请注明来源!

